

神经纤维瘤病虽然少见,但不可轻视。
Ⅰ型神经纤维瘤病(NF1)是一种常染色体显性遗传病,可引起皮肤、骨骼、脏器等的损伤和功能障碍,是目前医学诊疗的难点之一。
由于公众对该疾病认知的缺乏,部分医疗机构缺少NF1的诊疗经验,导致NF1群体容易被误诊、漏诊,错过最佳治疗时间,对患者身体造成无法挽回的损害。
不仅仅是皮肤表现,NF1可造成多方面危害。
神经纤维瘤病在临床上可分为3种主要类型:NF1、Ⅱ型神经纤维瘤病(NF2)和施万细胞瘤病,NF1是神经纤维瘤病的主要类型,其全球发病率约为1/3000,没有种族和性别差异。因其由von Recklinghausen在1882年首次描述,所以又被称为von Recklinghausen病,1987年美国国立卫生研究院(NIH)首次将其命名为NF1。
作为一种常染色体显性遗传病,NF1是由位于染色体17q11.2的NF1基因突变导致神经纤维蛋白失活或表达下调,从而产生以神经纤维瘤为主要特征的一系列神经皮肤损害,可合并各种良性和恶性肿瘤。NF1的临床症状常常表现为多发咖啡牛奶斑、雀斑、Lisch结节、骨骼畸形和认知功能障碍,甚至伴有危及生命的多发神经纤维瘤和其它肿瘤,其中多发咖啡牛奶斑和神经纤维瘤是NF1的标志性特征。
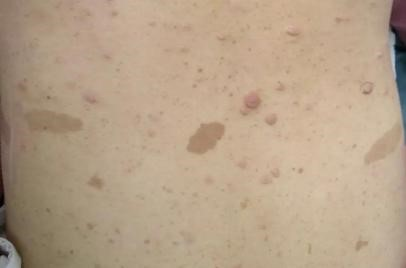
图1:牛奶咖啡斑
年龄不同,NF1患者的临床表现各异。
NF1临床特征的出现时间与严重程度因人而异,临床表现出现的典型顺序是咖啡牛奶斑、腋窝和(或)腹股沟雀斑、Lisch结节和神经纤维瘤。骨发育不良通常在出生后1年内出现,症状性视路胶质瘤通常到患者3岁时发生。其他肿瘤和神经系统并发症通常在患者出生1年后开始出现。高血压可能在儿童期出现,肿瘤恶变也可出现在儿童期,但更常发生于青春期和成年期。
在《Ⅰ型神经纤维瘤病临床诊疗专家共识(2021版)》中,也提出了中国NF1患者的不同临床表型自然史(图2:NF1的自然史流程图)。

图2:NF1的自然史流程图
NF1症状表现复杂多样,骨骼异常可包括脊柱侧弯及长骨发育不良,其中脊柱侧弯常出现在6~10岁或青春期早期。个人行为上的特征可包括认知障碍、注意力缺陷多动障碍(ADHD)和自闭症谱系障碍(ASD),同样多发于儿童期。肿瘤可包括恶性周围神经鞘瘤(MPNST)、视路胶质瘤和胶质瘤,前者在30岁左右最常见,但也可发生在任何年龄段。
丛状神经纤维瘤(pNF)是NF1的主要特征之一,表现为沿多个神经干和(或)分支形成大的肿瘤团块。pNF在较年轻患者中增长最快,但18岁以上患者中则很少出现每年肿瘤增长率超过20%的情况。大多数pNF伴随显著的临床症状,尤其是在早期易出现疼痛和运动功能障碍,肿瘤的迅速生长可伴随疼痛显著加重,而较大体积的肿瘤有更大可能导致运动功能障碍。
一般情况下,NF1可以通过体检和评估患者家族史来诊断。我国目前临床诊疗中NF1的诊断主要借鉴NIH于1987年共识会议上制定的临床标准,具体为:
(1)6个或以上咖啡牛奶斑:在青春期前直径>5mm或在青春期后直径>15mm;
(2)2个或以上任何类型的神经纤维瘤或1个pNF;
(3)腋窝或腹股沟区雀斑;
(4)视神经胶质瘤;
(5)2个或以上Lisch结节;
(6)特征性骨病变,如蝶骨发育不良或长骨皮质增厚伴或不伴假关节;
(7)有一级亲属(父母、同胞或子女)根据上述标准被诊断为NF1。
如果患者存在其中2种或以上的临床特征,可诊断为NF1。在2021年,国际神经纤维瘤病诊断标准共识组(I-NF-DC)对1987年制定的NF1诊断标准提出了修正建议,主要加入了基因学诊断内容,以进一步增加诊断的敏感度和准确性。
在诊断的同时需要注意需鉴别NF1与其它相似疾病,常见容易误诊的疾病有Legius综合征、McCune-Albright综合征、NF2、Noonan综合征、结构性错配修复缺陷综合征等,以下为常见相似疾病的临床特征(表1):

表1:NF1的鉴别诊断
牛奶咖啡斑和神经纤维瘤是NF1的显著特征,但NF1临床表现远不止这些,NF1可累及身体多个部位与系统(皮肤、神经、眼部、骨骼等),且症状表现轻重不一,给患者造成了包括容貌和身体功能等多方面影响,严重危害患者身心健康。加强对NF1的认知有利于NF1患者早诊断、早治疗,减少疾病给患者带来的多方面危害。








